文章图片
文章图片
文章图片
文章图片
文章图片

文章图片
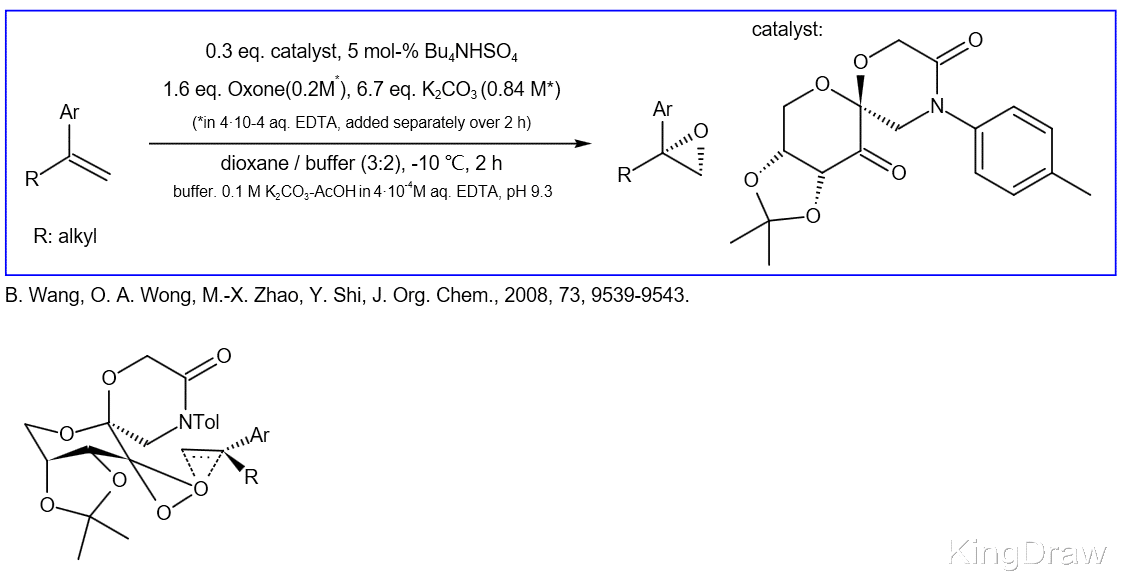
文章图片
史氏环氧化反应(Shi Epoxidation)
史氏环氧化反应 , 是华裔化学家史一安所发明的一种不对称环氧化反应 , 是指反式二取代的烯烃或三取代的烯烃在果糖衍生的手性酮催化下利用Oxone作为氧化剂进行不对称环氧化的反应 。 该反应的氧化剂是过一硫酸钾(KHSO5) , 催化剂是果糖的衍生物 。 史氏环氧化反应适用范围广泛 , 反式二取代烯与三取代烯均可作为反应的底物 , 因此成为有机合成的重要工具之一 。
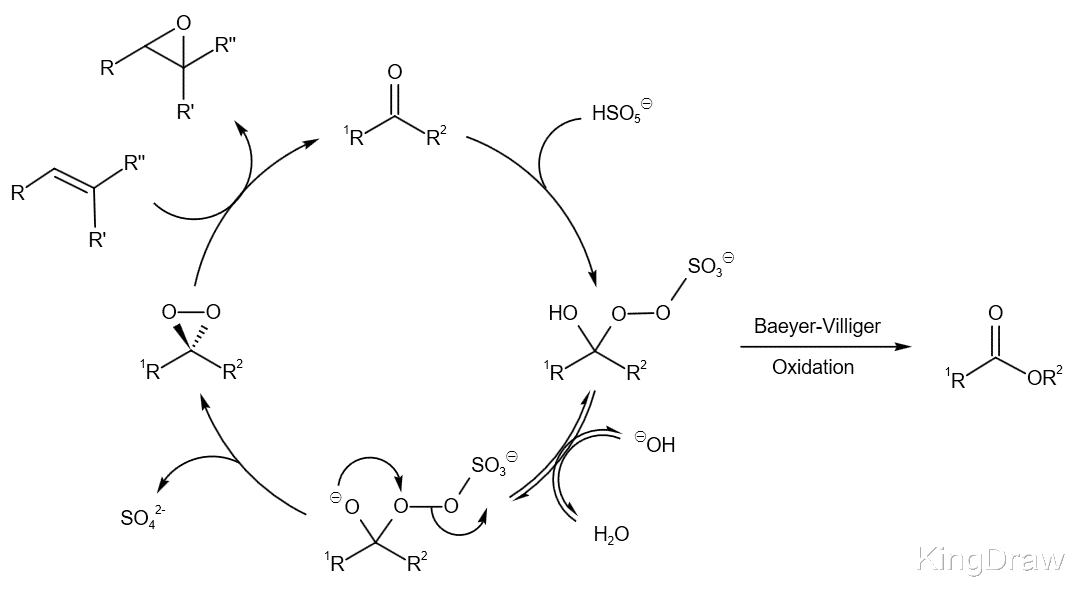
反应机理在反应中 , 真正的氧化剂是具有氧化性的被活化的手性过氧化酮 , 这个过氧酮的中间体是通过手性在原位被Oxone氧化而得到的 , 而这个中间体可以看做是过氧丙酮的类似物 , 之后这样的过氧丙酮类似物和烯烃发生环氧化反应得到环氧化物的产物 。
但是反应会存在一个竞争的反应 , 即反应中会发生Baeyer-Villiger重排 , 也就是Baeyer-Villiger氧化反应 , 这个副反应会严重的影响底物的转化率 。 当体系的pH从7上升到大于10时 , 反应的转化率可以提高十倍不止 , 但是反应的立体选择性的变化不是很大 , 约在92% e.e. 。 反应转化率的提升可能是因为副反应Baeyer-Villiger氧化反应减少的原因 , 同时增强了Oxone 的亲核性;但是在产生过氧化酮的过程中 , 对于pH的控制是至关重要的 , 因此史一安环氧化反应一般在pH 7-8进行 , 但是当反应体系的pH降低时 , 反应的效率会大幅度降低 , 因为过小的pH会导致Oxone发生自动分解反应 。
酮的反应性可以通过α位的吸电子基团来提高 。 从建立活性催化剂的早期尝试中得知 , 三氟甲基酮提高了活性 , 但是也可以使用其他吸电子基团 。 这些因素也降低了Bayer-Villiger氧化的速率 。 由于氢在α-位的酮易于外消旋化 , 因此手性元素经常被置于其他位置 。 这里显示了一些早期的催化剂:
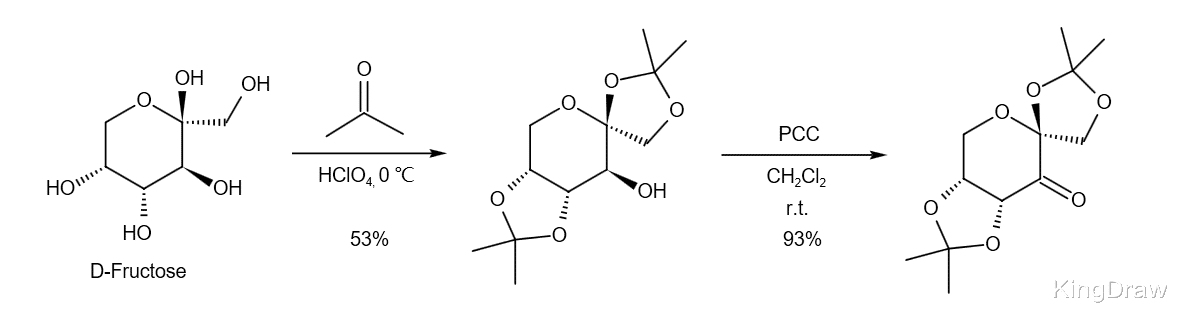
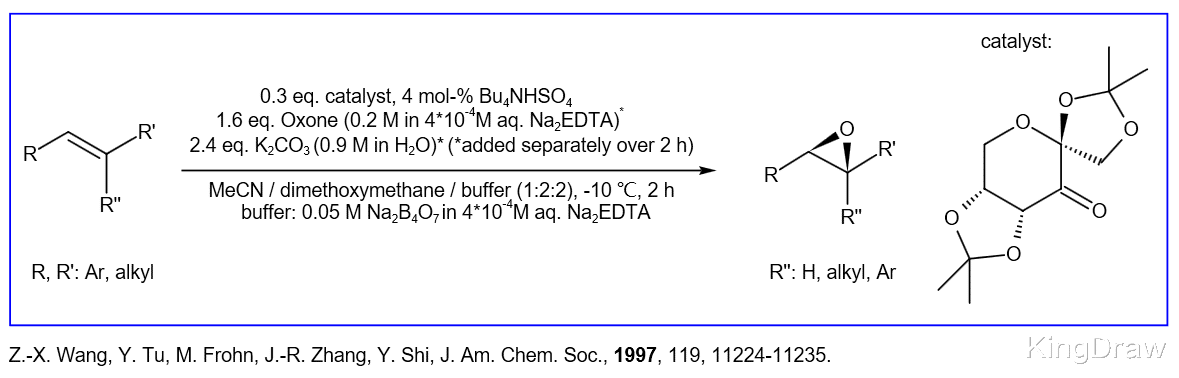
1996年 , 果糖衍生的酮被开发为高效的环氧化催化剂 。 该酮可以通过缩酮化和氧化从非常廉价的手性起始原料D-果糖分两步合成 。 由于可以从L-山梨糖合成L-果糖 , 因此该催化剂的对映异构体也可方便地获得 。
在这种催化剂中 , 立构中心靠近反应中心 , 因此底物和催化剂之间的立构化学连通是有效的 。 羰基的稠环或季中心α的存在使立体异构中心的差向异构化最小化 。 吸电子取代基活化羰基 。
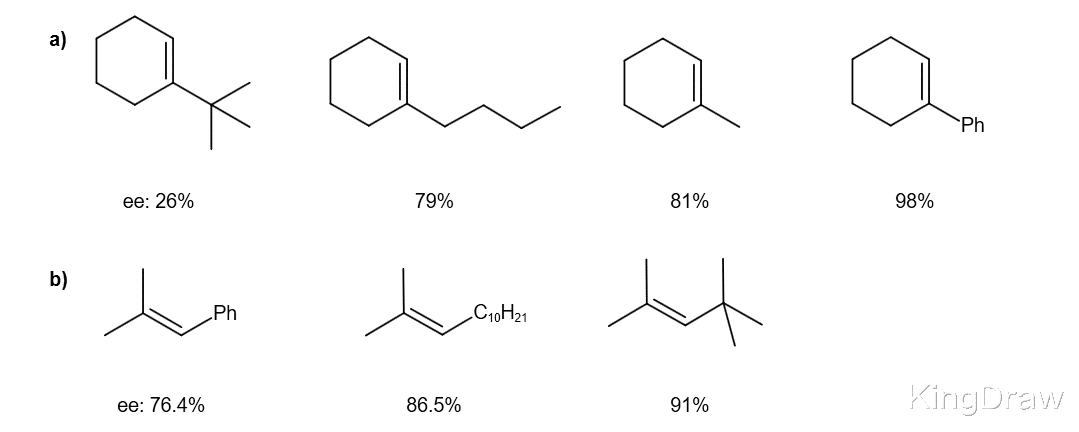
对于这种催化剂 , 在高于10的pH值下 , 反式-β-甲基苯乙烯的转化率比在较低的pH值(7-8)下提高了10倍 , 对映选择性仍然很高(90-92%) 。 这种引人注目的pH效应可使用催化量的酮 。 通过在反应进行时添加K2CO3可以方便地达到10.5的pH 。 该第一催化剂允许反式二取代和三取代的烯烃的高对映选择性转化 , 尽管对于顺式烯烃和末端烯烃而言 , 对映体过量仍然较低 。
由于稳定的氧孤对与烯烃的π*轨道的相互作用 , 螺环过渡态似乎是有利的 , 这在平面过渡态中是无法实现的 。
主要竞争模式是所示的平面过渡状态 。 如果R'体积较大(a) , 则三取代烯烃更有利 , 而较大的R取代基不利于平面过渡态(b) 。
后来的发展通过改变催化剂的取代方式实现了顺式取代的烯烃和端烯烃的转化 。 例如 , Boc保护的内酰胺允许顺式烯烃的转化 。
后来的发展通过改变催化剂的取代方式实现了顺式取代的烯烃和端烯烃的转化 。 例如 , Boc保护的内酰胺允许顺式烯烃的转化 。
在这里 , 可以假定具有π系统的基团与螺恶唑烷酮之间的相互作用 , 因此共轭苯乙烯和烯炔产生的对映体过量很高:
推荐阅读







- 地球的岩石,和月球的岩石有相同之处吗?
- ?了解更多关于俘获离子量子计算的详细信息
- 远隔38万公里,为何宇航员登上月球后,却偏偏不敢回头看地球?
- 私人公司也能训练宇航员,美国首个私人宇航员任务将于3月底发射
- 太空温度达零下270摄氏度,为什么太阳光到达地球后反而变热了?
- 在国际空间站中,人类可能受到的伤害
- 中国天眼收到外太空“警告”? 霍金生前或说对一件事!
- 1978年NASA发现金星生命,为何没公布,到底隐瞒了什么?
- 美俄真的做过太空受孕实验吗?失重状态下,对繁衍的影响多大?
- SpaceX火箭准备以每小时5000英里的速度撞击月球