我是斜杠青年 , 一个热爱前沿科学的“杂食性”学者!
模型无法同时准确预测许多蛋白质结构和分子的相互作用 。
来源:通过维基共享资源
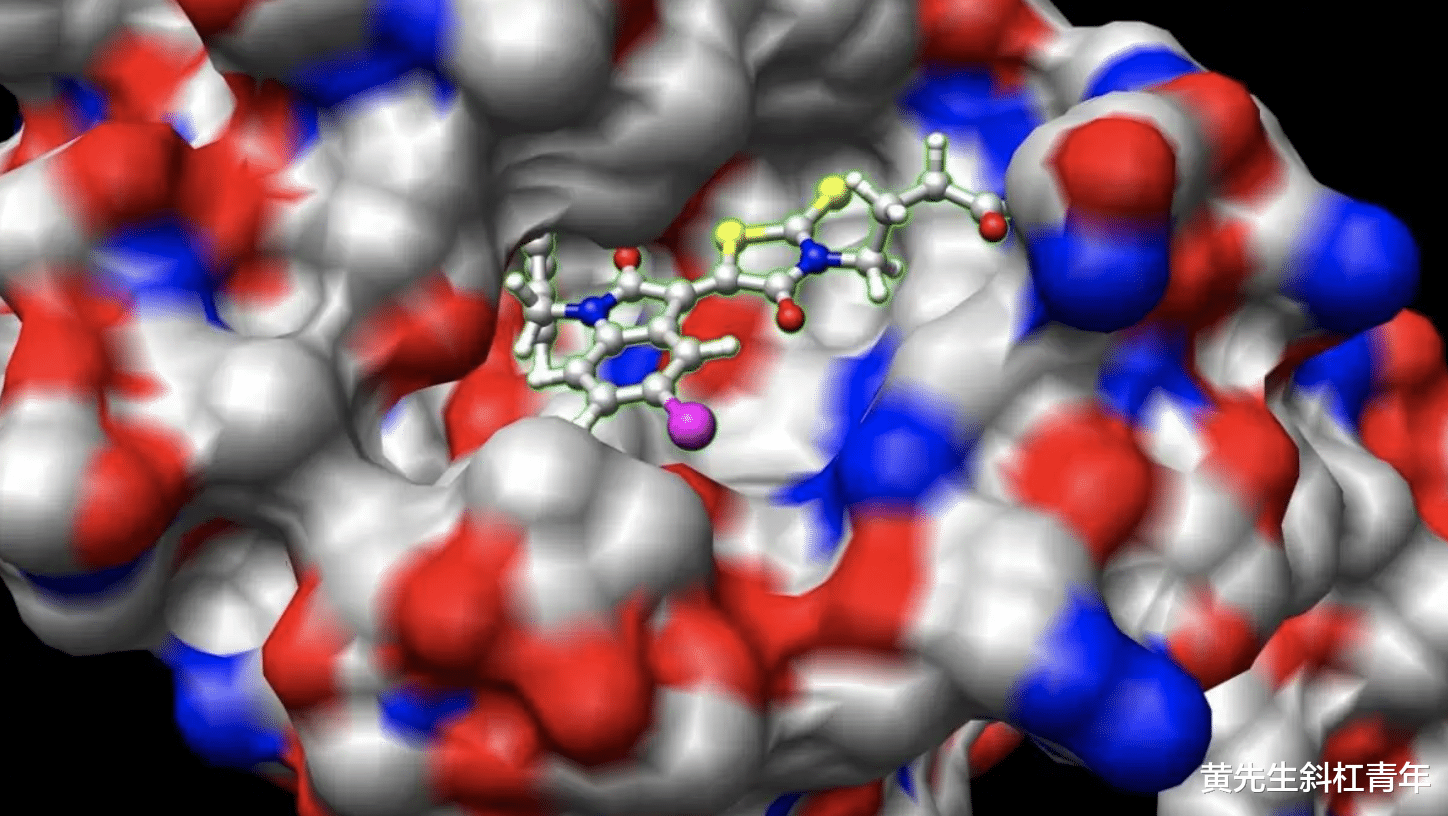
使用Chimera软件生成的分子表面Gold将化合物对接到蛋白质结构(乙酰转移酶) 。
科学家希望使用计算机模型来帮助降低与药物发现相关的成本和时间 , 开发新的抗生素来应对日益严重的抗菌素耐药性危机 。 但一项新的研究表明 , 目前一起使用最新工具并不比猜测的好多少 。
根据发表在分子系统生物学上的一项新研究 , 这是药物开发的障碍——至少在计算机模型现在存在的情况下 。
麻省理工学院(MIT)的研究人员探索了现有的计算机程序是否可以准确预测谷歌名为AlphaFold的新工具产生的抗菌化合物和细菌蛋白质结构之间的相互作用 , AlphaFold是一个人工智能程序 , 从其氨基酸序列生成3D蛋白质结构 。
令科学界兴奋的AlphaFold 。
但麻省理工学院团队发现 , 对现有模型的预测 , 即分子对接模拟 , 效果并不比预期的好 。
【现有的计算机模拟还不够强大,无法利用AlphaFold进行药物发现】麻省理工学院医学工程与科学研究所(IMES)和生物工程系医学工程和科学教授、高级作者James Collins表示:“AlphaFold等突破正在扩大硅胶(即通过计算机)药物发现工作的可能性 , 但这些发展需要与作为药物发现工作一部分的建模其他方面的额外进展相结合 。 ”
“该团队的研究既谈到了药物发现计算平台的当前能力和当前局限性 。 ”
希望科学家可以使用建模对影响以前未瞄准的细菌蛋白的新化合物进行大规模筛选 。 最终结果是开发了以前所未有的方式起作用的新抗生素 。
该团队使用分子对接模拟研究了大肠杆菌296种必需蛋白质与218种抗菌化合物的相互作用 , 该模拟根据它们的形状和物理特性预测两个分子的结合力 。
此前 , 这些模拟已被成功用于对照单个蛋白质靶点筛选大量化合物 , 以确定结合效果最好的化合物 。 但在这里 , 当试图根据许多潜在的蛋白质靶点筛选许多化合物时 , 预测变得不那么准确了 。
事实上 , 该模型在模拟现有药物与其目标之间的相互作用时产生了类似于真实阳性率的假阳性率 。
“利用这些标准的分子对接模拟 , 他们获得了大约0.5的auROC值 , 这基本上表明你做得并不比随机猜测好 。 ”
但这并不是因为AlphaFold的一些缺陷 , 因为当他们使用实验室实验确定的蛋白质结构相同的建模方法时 , 也发生了类似的结果 。
“AlphaFold似乎具有大致和实验确定的结构 , 但如果研究团队要在药物发现中有效和广泛地利用AlphaFold , 就需要更好地使用分子对接模型 。 ”
这种性能不佳的一个解释是 , 输入模型的蛋白质结构是静态的 , 但在真正的生物系统中 , 蛋白质是灵活的 , 并且经常改变其配置 。
研究人员通过运行四个额外的机器学习模型来提高分子对接模拟的性能 , 这些模型基于描述蛋白质和其他分子如何相互作用的数据进行训练 。
麻省理工学院柯林斯实验室的应用物理学家和博士后研究员、联合首席作者Felix Wong表示:“机器学习模型不仅学习了形状 , 还学习了已知相互作用的化学和物理性质 , 然后使用这些信息重新评估对接预测 。 ”
“他们发现 , 如果你使用这些额外的模型过滤交互 , 你可以获得更高的真实阳性与假阳性的比例 。 ”
“研究团队乐观地认为 , 随着建模方法的改进和计算能力的扩展 , 这些技术在药物发现中将变得越来越重要 。 ”“然而 , 要充分发挥硅药物发现的潜力 , 还有很长的路要走 。 ”
了解最新生命科学 , 关注我就是你最好的选择!
拓展阅读:
特斯拉Dojo架构大解析——道场微架构 - Hot Chips 34
新研究证实了1953年预测的“波纹片”蛋白质结构
推荐阅读







- 人类没有生殖隔离吗?为何黄种人、黑种人和白种人,能自由交配?
- 比黄金贵许多倍的元素,第一位一克上千亿
- 【未解之谜】人类身高不断增加之谜,你觉得这些说法哪个正确呢?
- 老家滑县到底怎么了?除了道口烧鸡,竟然因为疫情多发而出名了
- 时空穿越真的存在?男子还拿出了足够的证据!
- 重庆沙坪坝,一小区业主开车出去,发现自己的后车牌竟然被人戴上了口罩
- 女子在封控期间打麻将脑溢血死亡,棋牌室老板被判赔16万
- 科学家发现两个类星体即将碰撞,能量冲击波10000年后到达地球?